
 We work at the interface of data science and RNA biology/medicine, developing probabilistic models and machine learning algorithms to study RNA structure and function and applying these algorithms in large-scale genomic data analyses. One area of interest is transcriptome-wide analysis of post-transcriptional dynamics via integration of data from diverse high-throughput assays to infer sequence-structure-function relationships. RNA functions we study include RNA-protein interactions, gene expression, and stability or half-life (in vitro and in vivo). We are also interested in modeling the folding of RNA molecules using both biophysical models and high-throughput experiments, with the overarching goal of advancing the structure-driven discovery and engineering of novel RNAs for therapeutic and biotechnology applications.
We work at the interface of data science and RNA biology/medicine, developing probabilistic models and machine learning algorithms to study RNA structure and function and applying these algorithms in large-scale genomic data analyses. One area of interest is transcriptome-wide analysis of post-transcriptional dynamics via integration of data from diverse high-throughput assays to infer sequence-structure-function relationships. RNA functions we study include RNA-protein interactions, gene expression, and stability or half-life (in vitro and in vivo). We are also interested in modeling the folding of RNA molecules using both biophysical models and high-throughput experiments, with the overarching goal of advancing the structure-driven discovery and engineering of novel RNAs for therapeutic and biotechnology applications.
Transcriptomics |
|
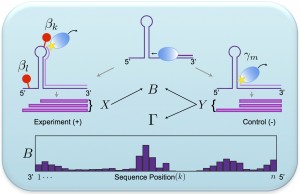 |
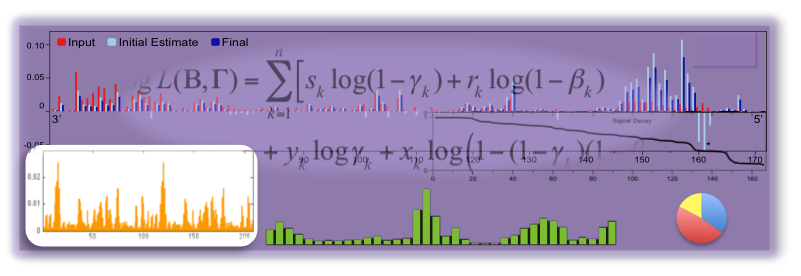
We develop and apply integrative data analysis methods for diverse sequencing-based assays, with particular emphasis on high-throughput measurements of RNA structure and RNA-protein interactions. Our work spans three aspects of data analysis: 1). statistically modeling experiments, their readouts, and the uncertainties in their data, 2). developing efficient and robust model-based inference algorithms to mine various quantities of interest from the data, and 3). analyzing these datasets in the context of other genomic datasets. |
RNA Sequence, Structure, and Function |
|
 |
We develop algorithms to infer predictive models of RNA folding and RNA function based on biological data and thermodynamic theory. This includes modeling the thermodynamic principles that underlie mRNA folding and the sequence and/or structure features that contribute to efficient protein binding, high gene expression, and slow mRNA decay. Better understanding of such sequence-structure-function relationships is fundamental to the design of highly functioning RNAs in a range of applications. |